
David L. Beveridge, PhD
Professor of Theoretical Chemistry and Molecular Biophysics
(860) 685-2575
|
|
David L. Beveridge, PhD Professor of Theoretical Chemistry and Molecular Biophysics (860) 685-2575 |
Theoretical physical chemistry and molecular biophysics, quantum mechanics, statistical thermodynamics, molecular dynamics and Monte Carlo computer simulation; structure, dynamics and solvation of nucleic acids and proteins, role of water in structural biochemistry.
Molecular Dynamics (MD) computer simulation is a powerful computational approach to the study of macromolecular structure and motions. MD simulation is a computer experiment in which the atoms of a postulated system execute Newtonian dynamics on an assumed potential energy surface. The DNA sequence chosen for study, the assumed energy surface, and the simulation protocol are all operational variables in the calculation. An accurate dynamical model for DNA sequences obtained from MD can in principle provide a general theoretical basis for understanding the nature of sequence-dependent structural irregularities, axis bending, and helical flexibility, all of which are expected to be implicated in important drug-DNA and protein-DNA interactions. This group specializes in MD simulations on DNA including explicit consideration of solvent, and validation of MD models of DNA via comparisons with experimental data. We are currently extending our considerations to the study of protein-DNA interactions. These studies are quite computationally intensive, and involve remote access to supercomputing facilities located at national resource centers as well as extensive use of local facilities for molecular graphics established on computer workstations at Wesleyan.
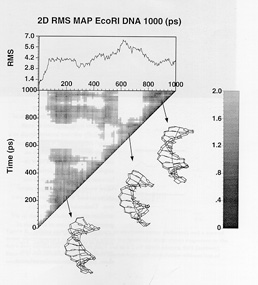 A
series of projects carried out recently have focused of developing a
dynamical model for the d(CGCGAATTCGCG) duplex, a system for which
considerable experimental data is available from x-ray
crystallography and nuclear magnetic resonance spectroscopy.
Particularly interesting structural deformations and axis bending
were observed in the crystal structure, and have formed the basis for
validation studies of the dynamical model of the sequence obtained
rom MD simulation. A comparison of calculated and observed NMR
parameters relevant to 2D--NOESY spectroscopy have served as a
validation point for the structure in solution. The MD results
provide a quite reasonable account of axis bending. These studies are
now being extended to DNA sequences involving sequences of adenine
residues, which have been previously associated with particular
bending characteristics in vitro and in nucleosome. A full scale
study of sequence effects on structure based on MD simulations is
envisaged.
A
series of projects carried out recently have focused of developing a
dynamical model for the d(CGCGAATTCGCG) duplex, a system for which
considerable experimental data is available from x-ray
crystallography and nuclear magnetic resonance spectroscopy.
Particularly interesting structural deformations and axis bending
were observed in the crystal structure, and have formed the basis for
validation studies of the dynamical model of the sequence obtained
rom MD simulation. A comparison of calculated and observed NMR
parameters relevant to 2D--NOESY spectroscopy have served as a
validation point for the structure in solution. The MD results
provide a quite reasonable account of axis bending. These studies are
now being extended to DNA sequences involving sequences of adenine
residues, which have been previously associated with particular
bending characteristics in vitro and in nucleosome. A full scale
study of sequence effects on structure based on MD simulations is
envisaged.
Most recently we have extended our MD studies into the realm of protein-DNA simulations, and developed a dynamical model for the repressor operator complex. Here we can study the uncomplexed DNA and protein independently as well as the complex, and use the results to investigate the role of sequence dependent effects, axis bending and deformability of the DNA on complex formation. These studies are designed to contribute to the eludcidation of the molecular basis of genetic regulatory mechanisms.
![]()
Selected Publications
![]()
Education
B.A. 1959 College of Wooster, Ohio
Ph.D. 1965 University of Cincinnati, Theoretical physical chemistry
Postdoctoral Fellowships:
Centre de Mécanique Ondulatoire Appliqué Paris, France;Carnegie Mellon University
 |
|
|
|
|
Last Updated: April 12, 1999